
Usuari:Jaumellecha/proves3
En matemàtiques, una funció de Green és la resposta a l'impuls d'un operador diferencial lineal no homogeni definit en un domini amb condicions inicials o condicions de contorn especificades.
Això vol dir que si L és l'operador diferencial lineal, aleshores
- la funció de Green G és la solució de l'equació LG = δ, on δ és la funció delta de Dirac;
- la solució del problema de valor inicial Ly = f és la convolució (G ⁎ f), on G és la funció de Green.
Mitjançant el principi de superposició, donada una equació diferencial ordinària lineal (EDO), L(solució) = font, primer es pot resoldre L(verd) = δs, per a cada s, i adonant-se que, ja que la font és una suma de funcions delta, la solució també és una suma de les funcions de Green, per linealitat de L.
-
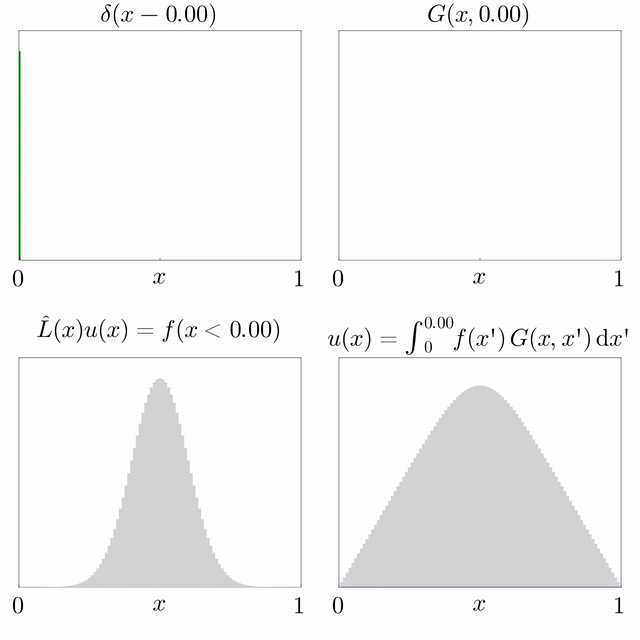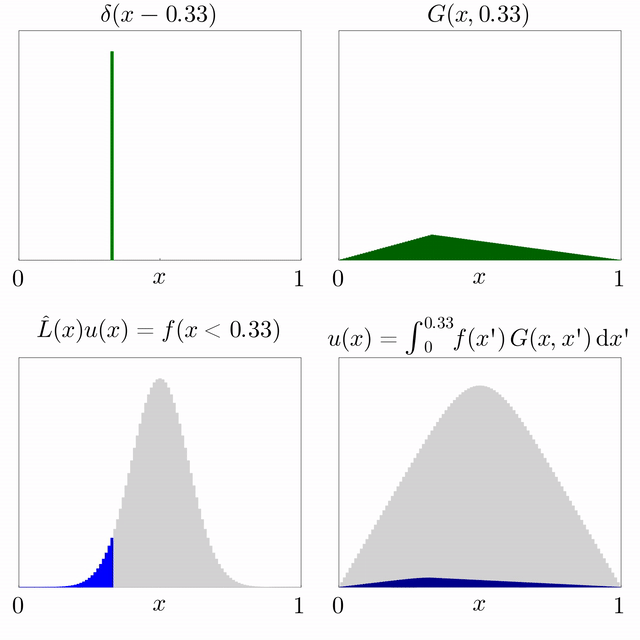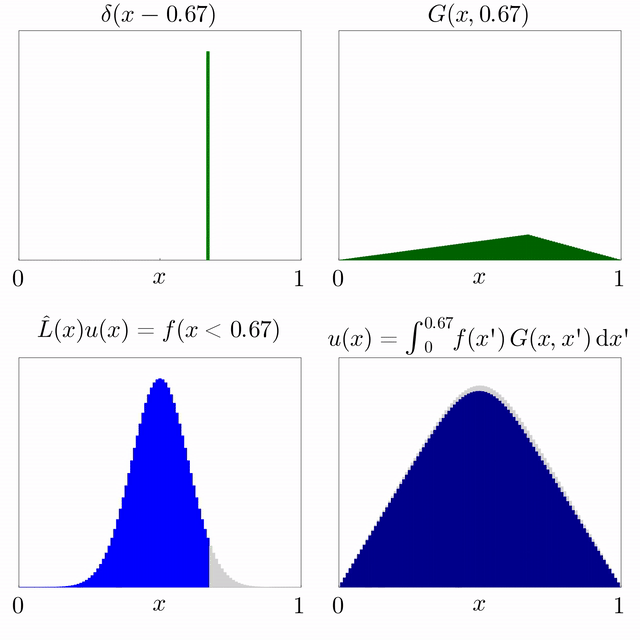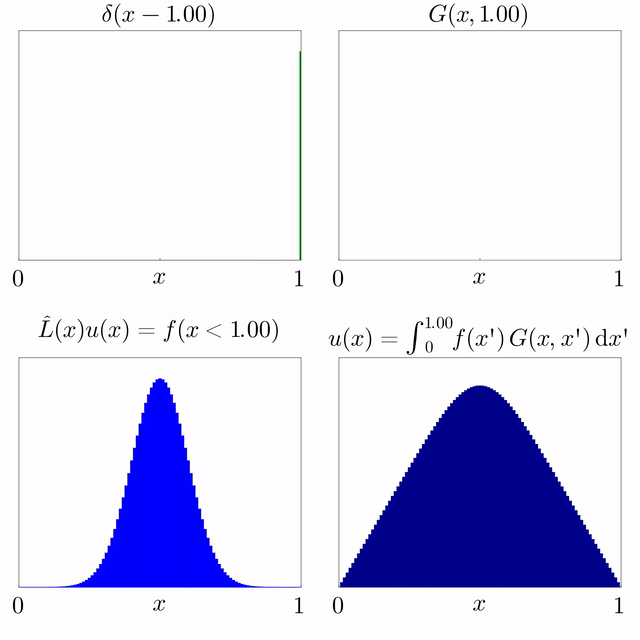 Si es coneix la solució d'una equació diferencial subjecta a una font puntual i l'operador diferencial és lineal, llavors es pot superposar per trobar la solució per a una font general
Si es coneix la solució d'una equació diferencial subjecta a una font puntual i l'operador diferencial és lineal, llavors es pot superposar per trobar la solució per a una font general





Les funcions de Green reben el nom del matemàtic britànic George Green, que va desenvolupar el concepte per primera vegada a la dècada del 1820. En l'estudi modern de les equacions diferencials en derivades parcials lineals, les funcions de Green s'estudien en gran part des del punt de vista de les solucions fonamentals.
Sota la teoria de molts cossos, el terme també s'utilitza en física, específicament en teoria quàntica de camps, aerodinàmica, aeroacústica, electrodinàmica, sismologia i teoria estadística de camps, per referir-se a diversos tipus de funcions de correlació, fins i tot aquelles que no s'ajusten a la definició matemàtica. En la teoria quàntica de camps, les funcions de Green prenen el paper de propagadors.
Definició i aplicacions
[modifica]A Green's function, G(x,s), of a linear differential operator acting on distributions over a subset of the Euclidean space , at a point s, is any solution of


|
|
() |

where δ is the Dirac delta function. This property of a Green's function can be exploited to solve differential equations of the form
|
|
() |

If the kernel of L is non-trivial, then the Green's function is not unique. However, in practice, some combination of symmetry, boundary conditions and/or other externally imposed criteria will give a unique Green's function. Green's functions may be categorized, by the type of boundary conditions satisfied, by a Green's function number. Also, Green's functions in general are distributions, not necessarily functions of a real variable.
Green's functions are also useful tools in solving wave equations and diffusion equations. In quantum mechanics, the Green's function of the Hamiltonian is a key concept with important links to the concept of density of states.
The Green's function as used in physics is usually defined with the opposite sign, instead. That is,This definition does not significantly change any of the properties of the Green's function due to the evenness of the Dirac delta function.

If the operator is translation invariant, that is, when has constant coefficients with respect to x, then the Green's function can be taken to be a convolution kernel, that is,


In this case, the Green's function is the same as the impulse response of linear time-invariant system theory.
Motivació
[modifica]Loosely speaking, if such a function G can be found for the operator , then, if we multiply the equation (1) for the Green's function by f(s), and then integrate with respect to s, we obtain,

Because the operator is linear and acts only on the variable x (and not on the variable of integration s), one may take the operator outside of the integration, yieldingThis means that

|
|
() |

is a solution to the equation

Thus, one may obtain the function u(x) through knowledge of the Green's function in equation (1) and the source term on the right-hand side in equation (2). This process relies upon the linearity of the operator .
In other words, the solution of equation (2), u(x), can be determined by the integration given in equation (3). Although f(x) is known, this integration cannot be performed unless G is also known. The problem now lies in finding the Green's function G that satisfies equation (1). For this reason, the Green's function is also sometimes called the fundamental solution associated to the operator .
Not every operator admits a Green's function. A Green's function can also be thought of as a right inverse of . Aside from the difficulties of finding a Green's function for a particular operator, the integral in equation (3) may be quite difficult to evaluate. However the method gives a theoretically exact result.
This can be thought of as an expansion of f according to a Dirac delta function basis (projecting f over ; and a superposition of the solution on each projection. Such an integral equation is known as a Fredholm integral equation, the study of which constitutes Fredholm theory.

Funcions de Green per a la resolució de problemes de valors de contorn no homogenis
[modifica]L'ús principal de les funcions de Green en matemàtiques és resoldre problemes de valors de contorn no homogenis. En la física teòrica moderna, les funcions de Green també s'utilitzen normalment com a propagadors en diagrames de Feynman; el terme funció de Green s'utilitza sovint per a qualsevol funció de correlació.
Framework
[modifica]Let be the Sturm–Liouville operator, a linear differential operator of the formand let be the vector-valued boundary conditions operator
![{\displaystyle \operatorname {L} ={\dfrac {d}{dx}}\left[p(x){\dfrac {d}{dx}}\right]+q(x)}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d52a1f66dc682885695c5e49068d6b8b3378e3ae)


Let be a continuous function in Further suppose that the problemis "regular", i.e., the only solution for for all x is .[a]

![{\displaystyle [0,\ell ]\,.}](https://wikimedia.org/api/rest_v1/media/math/render/svg/bc486e77bfe3c5b5be77a266bfa6707cb6f5732f)



Teorema
[modifica]There is one and only one solution that satisfiesand it is given bywhere is a Green's function satisfying the following conditions:



- is continuous in and .
- For , .
- For , .
- Derivative "jump": .
- Symmetry: .








Advanced and retarded Green's functions
[modifica]The Green's function is not necessarily unique since the addition of any solution of the homogeneous equation to one Green's function results in another Green's function. Therefore if the homogeneous equation has nontrivial solutions, multiple Green's functions exist. In some cases, it is possible to find one Green's functions that is nonvanishing only for , which is called a retarded Green's function, and another Green's function that is nonvanishing only for , which is called an advanced Green's function. In such cases any linear combination of the two Green's functions is also a valid Green's function. The terminology advanced and retarded is especially useful when the variable x corresponds to time. In such cases, the solution provided by the use of the retarded Green's function depends only on the past sources and is causal whereas the solution provided by the use of the advanced Green's function depends only on the future sources and is acausal. In these problems, it is often the case that the causal solution is the physically important one. The use of advanced and retarded Green's function is especially common for the analysis of solutions of the inhomogeneous electromagnetic wave equation.


Finding Green's functions
[modifica]Unitats
[modifica]While it doesn't uniquely fix the form the Green's function will take, performing a dimensional analysis to find the units a Green's function must have is an important sanity check on any Green's function found through other means. A quick examination of the defining equation,shows that the units of depend not only on the units of but also on the number and units of the space of which the position vectors and are elements. This leads to the relationship: where is defined as, "the physical units of ", and is the volume element of the space (or spacetime).



![{\displaystyle [[G]]=[[L]]^{-1}[[dx]]^{-1},}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4fdd0f71437271af1845d639fab3815f256b7ee4)
![{\displaystyle [[G]]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b9c4741f657ce5212f6d7e746b6db1174bb03569)

For example, if and time is the only variable then:If , the d'Alembert operator, and space has 3 dimensions then:

![{\displaystyle [[L]]=[[{\text{temps}}]]^{-2},}](https://wikimedia.org/api/rest_v1/media/math/render/svg/1b33df46b6f184c6f8de3e18934b5128519f342f)
![{\displaystyle [[dx]]=[[{\text{temps}}]],\ {\text{i}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/15fa7b41c5511fcf2a8b7f6b10352ca27a2b28e4)
![{\displaystyle [[G]]=[[{\text{temps}}]].}](https://wikimedia.org/api/rest_v1/media/math/render/svg/edc57b3cb42318d273e8733535f44eedb406594e)

![{\displaystyle [[L]]=[[{\text{longitud}}]]^{-2},}](https://wikimedia.org/api/rest_v1/media/math/render/svg/221bc103c929d724e58ab52762bde0aa49daa41e)
![{\displaystyle [[dx]]=[[{\text{temps}}]][[{\text{longitud}}]]^{3},\ {\text{i}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/af20b511e7105f40abd1a4a98e403745aca5e41a)
![{\displaystyle [[G]]=[[{\text{temps}}]]^{-1}[[{\text{longitud}}]]^{-1}.}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b9819a2b9e6f4c61ace802b5029e3d41ade1b649)
Eigenvalue expansions
[modifica]If a differential operator L admits a set of eigenvectors Ψn(x) (i.e., a set of functions Ψn and scalars λn such that LΨn = λn Ψn ) that is complete, then it is possible to construct a Green's function from these eigenvectors and eigenvalues.
"Complete" means that the set of functions Plantilla:Mset satisfies the following completeness relation,

Then the following holds,

where represents complex conjugation.

Applying the operator L to each side of this equation results in the completeness relation, which was assumed.
The general study of the Green's function written in the above form, and its relationship to the function spaces formed by the eigenvectors, is known as Fredholm theory.
There are several other methods for finding Green's functions, including the method of images, separation of variables, and Laplace transforms.[1]
Combining Green's functions
[modifica]If the differential operator can be factored as then the Green's function of can be constructed from the Green's functions for and :The above identity follows immediately from taking to be the representation of the right operator inverse of , analogous to how for the invertible linear operator , defined by , is represented by its matrix elements .







A further identity follows for differential operators that are scalar polynomials of the derivative, . The fundamental theorem of algebra, combined with the fact that commutes with itself, guarantees that the polynomial can be factored, putting in the form:where are the zeros of . Taking the Fourier transform of with respect to both and gives:The fraction can then be split into a sum using a partial fraction decomposition before Fourier transforming back to and space. This process yields identities that relate integrals of Green's functions and sums of the same. For example, if then one form for its Green's function is:While the example presented is tractable analytically, it illustrates a process that works when the integral is not trivial (for example, when is the operator in the polynomial).








![{\displaystyle {\begin{aligned}G(x,s)&={\frac {1}{(\alpha -\gamma )^{2}}}\Theta (x-s)e^{-\gamma (x-s)}-{\frac {1}{(\alpha -\gamma )^{2}}}\Theta (x-s)e^{-\alpha (x-s)}+{\frac {1}{\gamma -\alpha }}\Theta (x-s)\,(x-s)e^{-\alpha (x-s)}\\[5pt]&=\int \Theta (x-s_{1})(x-s_{1})e^{-\alpha (x-s_{1})}\Theta (s_{1}-s)e^{-\gamma (s_{1}-s)}\,ds_{1}.\end{aligned}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e6ac1f0afa2d7a8139363f65c34e4666f515ba16)

Taula de les funcions de Green
[modifica]The following table gives an overview of Green's functions of frequently appearing differential operators, where , , is the Heaviside step function, is a Bessel function, is a modified Bessel function of the first kind, and is a modified Bessel function of the second kind.[2] Where time (t) appears in the first column, the advanced (causal) Green's function is listed.






| Operador diferencial L | Funció de Green G | Exemple d'aplicació |
|---|---|---|
| on | amb | 1D underdamped harmonic oscillator |
| on | amb | 1D overdamped harmonic oscillator |
| on | 1D critically damped harmonic oscillator | |
| Operador laplacià 2D | amb | 2D Poisson equation |
| Operador laplacià 3D | amb | Poisson equation |
| Operador de Helmholtz | stationary 3D Schrödinger equation for free particle | |
| en dimensions | Yukawa potential, Feynman propagator | |
| 1D wave equation | ||
| 2D wave equation | ||
| Operador de D'Alembert | 3D wave equation | |
| 1D diffusion | ||
| 2D diffusion | ||
| 3D diffusion | ||
| amb | 1D Klein–Gordon equation | |
| amb | 2D Klein–Gordon equation | |
| amb | 3D Klein–Gordon equation | |
| amb | telegrapher's equation | |
| amb | 2D relativistic heat conduction | |
| amb | 3D relativistic heat conduction |










































![{\displaystyle {\frac {1}{2}}\left[\left(1-\sin {\mu ct}\right)(\delta (ct-x)+\delta (ct+x))+\mu \Theta (ct-|x|)J_{0}(\mu u)\right]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c44899b6b62879a8f6959a6510d4b8ec068de259)


![{\displaystyle {\frac {1}{4\pi }}\left[(1+\cos(\mu ct)){\frac {\delta (ct-\rho )}{\rho }}+\mu ^{2}\Theta (ct-\rho )\operatorname {sinc} (\mu u)\right]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b757b456e58fb871ce7bb29585a38f85ddce2612)


![{\displaystyle {\frac {1}{4\pi }}\left[{\frac {\delta \left(t-{\frac {r}{c}}\right)}{r}}+\mu c\Theta (ct-r){\frac {J_{1}\left(\mu u\right)}{u}}\right]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/490b291c01f8db1ae5d862628c52b911edcfb68a)


![{\displaystyle {\frac {1}{2}}e^{-\gamma t}\left[\delta (ct-x)+\delta (ct+x)+\Theta (ct-|x|)\left({\frac {\gamma }{c}}I_{0}\left({\frac {\gamma u}{c}}\right)+{\frac {\gamma t}{u}}I_{1}\left({\frac {\gamma u}{c}}\right)\right)\right]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/2a1b46c106358dc97a87804ca03bc389342c9fcd)

![{\displaystyle {\frac {e^{-\gamma t}}{4\pi }}\left[(1+e^{-\gamma t}+3\gamma t){\frac {\delta (ct-\rho )}{\rho }}+\Theta (ct-\rho )\left({\frac {\gamma \sinh \left({\frac {\gamma u}{c}}\right)}{cu}}+{\frac {3\gamma t\cosh \left({\frac {\gamma u}{c}}\right)}{u^{2}}}-{\frac {3ct\sinh \left({\frac {\gamma u}{c}}\right)}{u^{3}}}\right)\right]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/2fc047f78947bea8529e1911ba3963aed2bbd06f)

![{\displaystyle {\frac {e^{-\gamma t}}{20\pi }}\left[\left(8-3e^{-\gamma t}+2\gamma t+4\gamma ^{2}t^{2}\right){\frac {\delta (ct-r)}{r^{2}}}+{\frac {\gamma ^{2}}{c}}\Theta (ct-r)\left({\frac {1}{cu}}I_{1}\left({\frac {\gamma u}{c}}\right)+{\frac {4t}{u^{2}}}I_{2}\left({\frac {\gamma u}{c}}\right)\right)\right]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/dd7444bb46d1bfc9f243acfa196ffc3dc8843030)
Green's functions for the Laplacian
[modifica]Green's functions for linear differential operators involving the Laplacian may be readily put to use using the second of Green's identities.
To derive Green's theorem, begin with the divergence theorem (otherwise known as Gauss's theorem),

Let and substitute into Gauss' law.

Compute and apply the product rule for the ∇ operator,


Plugging this into the divergence theorem produces Green's theorem,

Suppose that the linear differential operator L is the Laplacian, ∇2, and that there is a Green's function G for the Laplacian. The defining property of the Green's function still holds,

Let in Green's second identity, see Green's identities. Then,

![{\displaystyle \int _{V}\left[\varphi (x')\delta (x-x')-G(x,x')\,{\nabla '}^{2}\,\varphi (x')\right]\ d^{3}x'=\int _{S}\left[\varphi (x')\,{\nabla '}G(x,x')-G(x,x')\,{\nabla '}\varphi (x')\right]\cdot d{\widehat {\sigma }}'.}](https://wikimedia.org/api/rest_v1/media/math/render/svg/2a6deb7207fd88c9f116a786927a3b2410821523)
Using this expression, it is possible to solve Laplace's equation ∇2φ(x) = 0 or Poisson's equation ∇2φ(x) = −ρ(x), subject to either Neumann or Dirichlet boundary conditions. In other words, we can solve for φ(x) everywhere inside a volume where either (1) the value of φ(x) is specified on the bounding surface of the volume (Dirichlet boundary conditions), or (2) the normal derivative of φ(x) is specified on the bounding surface (Neumann boundary conditions).
Suppose the problem is to solve for φ(x) inside the region. Then the integral reduces to simply φ(x) due to the defining property of the Dirac delta function and we have

![{\displaystyle \varphi (x)=-\int _{V}G(x,x')\rho (x')\ d^{3}x'+\int _{S}\left[\varphi (x')\,\nabla 'G(x,x')-G(x,x')\,\nabla '\varphi (x')\right]\cdot d{\widehat {\sigma }}'.}](https://wikimedia.org/api/rest_v1/media/math/render/svg/527d0f78422408b1f342eedff063e16a96162e26)
This form expresses the well-known property of harmonic functions, that if the value or normal derivative is known on a bounding surface, then the value of the function inside the volume is known everywhere.
In electrostatics, φ(x) is interpreted as the electric potential, ρ(x) as electric charge density, and the normal derivative as the normal component of the electric field.

If the problem is to solve a Dirichlet boundary value problem, the Green's function should be chosen such that G(x,x′) vanishes when either x or x′ is on the bounding surface. Thus only one of the two terms in the surface integral remains. If the problem is to solve a Neumann boundary value problem, the Green's function is chosen such that its normal derivative vanishes on the bounding surface, as it would seem to be the most logical choice. (See Jackson J.D. classical electrodynamics, page 39). However, application of Gauss's theorem to the differential equation defining the Green's function yieldsmeaning the normal derivative of G(x,x′) cannot vanish on the surface, because it must integrate to 1 on the surface. (Again, see Jackson J.D. classical electrodynamics, page 39 for this and the following argument).

The simplest form the normal derivative can take is that of a constant, namely 1/S, where S is the surface area of the surface. The surface term in the solution becomeswhere is the average value of the potential on the surface. This number is not known in general, but is often unimportant, as the goal is often to obtain the electric field given by the gradient of the potential, rather than the potential itself.


With no boundary conditions, the Green's function for the Laplacian (Green's function for the three-variable Laplace equation) is

Supposing that the bounding surface goes out to infinity and plugging in this expression for the Green's function finally yields the standard expression for electric potential in terms of electric charge density as

Example
[modifica]Find the Green function for the following problem, whose Green's function number is X11:

First step: The Green's function for the linear operator at hand is defined as the solution to
|
|
(Eq. ) |

If , then the delta function gives zero, and the general solution is


For , the boundary condition at implies



if and .

For , the boundary condition at implies



The equation of is skipped for similar reasons.

To summarize the results thus far:

Second step: The next task is to determine and .


Ensuring continuity in the Green's function at implies


One can ensure proper discontinuity in the first derivative by integrating the defining differential equation (i.e., Eq. *) from to and taking the limit as goes to zero. Note that we only integrate the second derivative as the remaining term will be continuous by construction.




The two (dis)continuity equations can be solved for and to obtain

So the Green's function for this problem is:

Further examples
[modifica]- Let n = 1 and let the subset be all of R. Let L be . Then, the Heaviside step function H(x − x0) is a Green's function of L at x0.
- Let n = 2 and let the subset be the quarter-plane {(x, y) : x, y ≥ 0} and L be the Laplacian. Also, assume a Dirichlet boundary condition is imposed at x = 0 and a Neumann boundary condition is imposed at y = 0. Then the X10Y20 Green's function is
- Let , and all three are elements of the real numbers. Then, for any function with an -th derivative that is integrable over the interval : The Green's function in the above equation, , is not unique. How is the equation modified if is added to , where satisfies for all (for example, with )? Also, compare the above equation to the form of a Taylor series centered at .

![{\displaystyle {\begin{aligned}G(x,y,x_{0},y_{0})={\dfrac {1}{2\pi }}&\left[\ln {\sqrt {(x-x_{0})^{2}+(y-y_{0})^{2}}}-\ln {\sqrt {(x+x_{0})^{2}+(y-y_{0})^{2}}}\right.\\[5pt]&\left.{}+\ln {\sqrt {(x-x_{0})^{2}+(y+y_{0})^{2}}}-\ln {\sqrt {(x+x_{0})^{2}+(y+y_{0})^{2}}}\,\right].\end{aligned}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/13fbf122af36e07bc209fd6fa423687e8d8f63c1)


![{\displaystyle [a,b]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9c4b788fc5c637e26ee98b45f89a5c08c85f7935)
![{\displaystyle {\begin{aligned}f(x)&=\sum _{m=0}^{n-1}{\frac {(x-a)^{m}}{m!}}\left[{\frac {d^{m}f}{dx^{m}}}\right]_{x=a}+\int _{a}^{b}\left[{\frac {(x-s)^{n-1}}{(n-1)!}}\Theta (x-s)\right]\left[{\frac {d^{n}f}{dx^{n}}}\right]_{x=s}ds\end{aligned}}~.}](https://wikimedia.org/api/rest_v1/media/math/render/svg/fc9ecbd68e5ee3907569c9d36bb93634bef1aaf4)




![{\displaystyle x\in [a,b]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/026357b404ee584c475579fb2302a4e9881b8cce)



Notes
[modifica]- ↑ In technical jargon "regular" means that only the trivial solution () exists for the homogeneous problem ().
Referències
[modifica]Bibliogragia
[modifica]- Bayin, S. S. Mathematical Methods in Science and Engineering (en anglès). Wiley, 2006, p. capítol 18 i 19.
- Cole, K. D; Beck, J. V; Haji-Sheikh, A; Litkouhi, B. «Methods for obtaining Green's functions». A: Heat Conduction Using Green's Functions (en anglès). Taylor and Francis, 2011, p. 101-148. ISBN 978-1-4398-1354-6.
- Eyges, Leonard. The Classical Electromagnetic Field (en anglès). Nova York, NY: Dover Publications, 1972. ISBN 0-486-63947-9. El capítol 5 conté una explicació molt llegible de l'ús de les funcions de Green per resoldre problemes de valors de límit en electrostàtica.
- Faryad, M; Lakhtakia, A. Infinite-Space Dyadic Green Functions in Electromagnetism (en anglès). Londres, UK / San Rafael, CA: IoP Science (UK) / Morgan and Claypool (US), 2018.
- Folland, G. B. Fourier Analysis and its Applications (en anglès). Wadsworth and Brooks/Cole (Mathematics Series).
- Green, G. An Essay on the Application of Mathematical Analysis to the Theories of Electricity and Magnetism (en anglès). Nottingham, Anglaterra: T. Wheelhouse, 1828, p. 10-12.
- Mathews, Jon; Walker, Robert L. Mathematical methods of physics (en anglès). Nova York: W. A. Benjamin, 1970. ISBN 0-8053-7002-1.
- Polyanin, A. D. Handbook of Linear Partial Differential Equations for Engineers and Scientists (en anglès). Boca Raton, FL: Chapman & Hall/CRC Press, 2002. ISBN 1-58488-299-9.
- Polyanin, A. D; Zaitsev, V. F. Handbook of Exact Solutions for Ordinary Differential Equations (en anglès). Boca Raton, FL: Chapman & Hall/CRC Press, 2003.
- Schulz, Hermann. Physik mit Bleistift (en alemany), 2001.
Vegeu també
[modifica]- Bessel potential
- Discrete Green's functions – defined on graphs and grids
- Impulse response – the analog of a Green's function in signal processing
- Transfer function
- Fundamental solution
- Green's function in many-body theory
- Correlation function
- Propagator
- Green's identities
- Parametrix
- Volterra integral equation
- Resolvent formalism
- Keldysh formalism
- Spectral theory
Enllaços externs
[modifica]- Michiel Hazewinkel (ed.). p/g045090. Encyclopedia of Mathematics (en anglès). Springer, 2001. ISBN 978-1-55608-010-4.
- Weisstein, Eric W., «Green's Function» a MathWorld (en anglès).
- Green's function for differential operator a PlanetMath
- Green's function a PlanetMath
- Green functions and conformal mapping a PlanetMath
- Jauho, A. P. «Introduction to the Keldysh Nonequilibrium Green Function Technique» (en anglès). Nanohub.
- «Green's Function Library» (en anglès).
- «Tutorial on Green's functions» (en anglès).
- «Boundary Element Method (for some idea on how Green's functions may be used with the boundary element method for solving potential problems numerically)» (en anglès).
- «Green's function» (en anglès).
- «Delta function and Green's function» (en anglès). MIT video lecture on Green's function.
- Bowley, Roger. «George Green & Green's Functions» (en anglès).}}