
Enllaç C-Au
Els compostos d'organoor (o compostos orgànics de l'or) són compostos químics que contenen un enllaç químic entre carboni (C) i or (Au) (enllaç C-Au). La química de l'organoor és la química dels compostos organometàl·lics que contenen un enllaç químic carboni-or i descriu les propietats físiques, la síntesi, les reaccions i l'ús d'aquests compostos.
S'estudien en recerca acadèmica, però no han rebut un ús generalitzat d'una altra manera. Els estats d'oxidació dominants per als compostos organoor són I amb el nombre de coordinació 2 i una geometria molecular lineal i III amb CN = 4 i una geometria molecular quadrada plana.[1][2][3] El primer compost d'organoor descobert va ser el carbur d'or (I) Au₂C₂, que es va preparar per primera vegada l'any 1900.[4]
Or(I)[modifica]
Els complexos d'or (I) són espècies de 14 electrons, lineals, diamagnètics i de 2 coordenades.[1][2][3] Normalment existeixen com a adductes LAuR amb com a lligand L, per exemple, una trifenilfosfina o un isocianur. El lligand impedeix la reducció de Au(I) a Au(0) metàl·lic amb la dimerització del residu orgànic. L'or (I) també pot existir com l'aurat M[AuR₂] (el complex at), per la qual cosa el catió sol estar equipat amb un agent complexant per millorar l'estabilitat. L'anió AuR₂− també és lineal igual que altres espècies M(d10) com Hg(Me)₂ i Pd(Me)₂2+. Se sap que l'or forma acetilurs (capaços de formar estructures polimèriques), carbens i carbins. El mètode clàssic per a la preparació de compostos LAuR és per reacció d'un reactiu de Grignard amb un halogenur d'or(I). Una reacció posterior amb un organoliti R-Li forma el complex at.
En un grup especial de compostos, un àtom de carboni aril actua com a pont entre dos àtoms d'or. Un d'aquests compostos, (MesAu)5, es forma en una reacció entre Au(CO)Cl i el mesitil Grignard. El carboni es pot coordinar amb l'or fins a un valor de 6. Els compostos del tipus C(AuL)4 són isolobal amb metà i els de tipus C(AuL)5+ isolòbals amb l'ió metà. Aquests cúmuls d'or hipercoordinats sovint s'estabilitzen per interaccions auròfiles entre els centres d'or formalment tancats.[5]
-
 Algunes espècies típiques d'organoor amb diferents modes d'enllaç
Algunes espècies típiques d'organoor amb diferents modes d'enllaç

Els compostos de cianur d'or (MAu(CN)₂) tenen certa importància per a la cianuració de l'or, un procés per a l'extracció d'or del mineral de baixa qualitat. L'enllaç carboni-metall en els cianurs metàl·lics sol ser iònic, però hi ha proves que l'enllaç C-Au en l'ió cianur d'or és covalent.[6]
Or(III)[modifica]
Els complexos d'or(III) són 4 coordenades, quadrades, diamagnètiques, tòxiques, 16 espècies d'electrons. Quan el nombre de coordinació formal és inferior a 4, lligands com el clor ho poden compensar formant un lligand de pont. La quelació intramolecular és una altra estratègia. En general, els compostos d'or(III) són tòxics i, per tant, menys estudiats que l'or(I). Els complexos monoarilor(III) són una classe de complexos ben estudiada. Sovint es preparen mitjançant l'auració electròfila directa d'arens per AuCl₃.[7]
Els complexos de tetraalquilaurat (III) homolèptics (per exemple, Li[AuMe4]) també estan ben caracteritzats.[8]
Catàlisi de l'or[modifica]
Consideracions generals[modifica]
Les reaccions catalitzades per or es divideixen en dues categories principals: catàlisi heterogènia que inclou catalitzadors per nanopartícules d'or (per exemple, Au/TiO₂) i superfícies d'or de monocapa de tiol, i catalitzadors sobre suport d'alúmina, inclosa Au/CeO₂ suportada per alúmina. Aquests catalitzadors s'han investigat per a processos industrialment importants com l'oxidació d'alcohols, l'oxidació de monòxid de carboni (CO) i diverses reaccions d'hidrogenació selectiva (per exemple, butadiè a butè). Tot i que sovint és eficient i presenta selectivitats útils o úniques, hi ha una incertesa considerable pel que fa al mecanisme dels processos catalitzats per diversos catalitzadors d'or heterogenis, fins i tot en comparació amb altres catalitzadors heterogenis de metalls de transició.
En canvi, la catàlisi homogènia amb or utilitza compostos d'or(I) o or(III) simples o units a lligands que són solubles en dissolvents orgànics i s'utilitza per a la síntesi de productes químics fins en química orgànica.[9][10] Els halogenurs d'or binaris i els complexos simples, inclosos el clorur d'or(I), el clorur d'or(III) i l'àcid cloroàuric, s'han utilitzat com a complexos. Però aquestes fonts d'or donen lloc ràpidament a catalitzadors actius en solució mal definits i fàcilment desactivables (mitjançant la reducció a Au(0)). El desenvolupament de complexos d'or(I) lligats amb fosfina o NHC ben definits va ser un avenç important i va provocar un augment significatiu de l'interès en les aplicacions sintètiques de la catàlisi de l'or. Els complexos d'or(I) lligats normalment es preparen i emmagatzemen com a clorurs estables al banc (però poc reactius), LAuCl, per exemple, cloro(trifenilfosfina)or(I), que normalment s'activen mitjançant l'abstracció d'halogenurs amb sals de plata com AgOTf, AgBF4, o AgSbF6 per generar una espècie d'or(I) catiònic.[11][12] Tot i que el complex coordinativament insaturat «LAu+» es genera a partir d'una barreja LAuCl/AgX; la naturalesa exacta de l'espècie d'or catiònic i el paper de la sal de plata segueix sent una mica controvertida.[13][14][15] El para-nitrobenzoat, la bistriflimida i certs complexos de nitril representen precatalitzadors sense plata catalíticament actius però aïllables.
L'or(I) catiònic forma complexos π amb enllaços alquens o alquins, seguint el model Dewar-Chatt-Duncanson. Certament, l'or no és l'únic metall que mostra aquest tipus d'enllaç i reactivitat, també ho fan diversos ions metàl·lics isolobals amb el protó simple (és a dir, un orbital s buit): per exemple, el mercuri(II) i el platí(II). Els ions electròfils i els complexos com aquests amb una forta propensió a formar complexos π es coneixen generalment com àcids pi(π) (vegeu també: Interacció catió-π).[16]
-
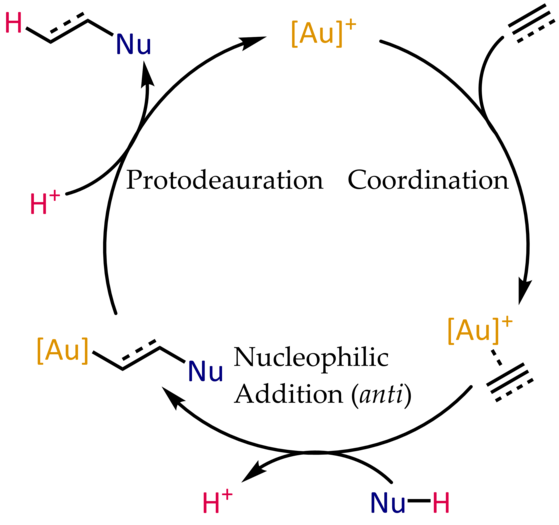 Mecanisme típic per a la hidrofuncionalització catalitzada per or(I) d'alquins i al·lèns
Mecanisme típic per a la hidrofuncionalització catalitzada per or(I) d'alquins i al·lèns

Els complexos d'or(I)-alqué i -alquí són electròfils i susceptibles a l'atac nucleòfil. En l'oximercuració, l'espècie organomercurial resultant es genera estequiomètricament i requereix un pas addicional per alliberar el producte. En el cas de l'or, la protonòlisi de l'enllaç Au-C tanca el cicle catalític, permetent la coordinació d'un altre substrat.
Alguns avantatges pràctics de la catàlisi d'or(I) inclouen:
1) estabilitat de l'aire (a causa de l'alt potencial d'oxidació de l'Au(I)),
2) tolerància a la humitat adventícia (a causa de la seva baixa oxofilia) i
3) toxicitat relativament baixa en comparació amb altres pi-àcids (per exemple, Pt(II) i Hg(II)).
Químicament, els complexos Au(I) normalment no experimenten oxidació a estats d'oxidació més alts, i els Au(I)-alquils i -vinils no són susceptibles a l'eliminació d'hidrur β.[17]
Història del seu desenvolupament[modifica]
El 1976, Thomas i els seus companys van informar de la conversió de fenilacetilè a acetofenona utilitzant àcid tetracloroàuric amb un rendiment del 37%.[18] En aquesta reacció es va utilitzar l'or(III) com a catalitzador homogeni substituint el mercuri en l'oximercuració. Aquest mateix estudi enumera un rendiment publicat >150%, indicant una catàlisi que potser no va ser reconeguda pels químics.
El 1991, Utimoto va fer reaccionar l'or(III) (NaAuCl4) amb alquins i aigua.[19] Teles va identificar un inconvenient important d'aquest mètode, ja que Au(III) es va reduir ràpidament a or metàl·lic mort catalíticament i el 1998 va tornar al tema de l'Au(I) suportat per lligands per a la mateixa transformació:[20]

Aquesta reacció en particular va demostrar una eficiència catalítica fantàstica i desencadenaria una sèrie d'investigacions sobre l'ús de complexos de fosfingol(I) per a l'activació de múltiples enllaços C-C en els propers anys.[21] Malgrat la menor estabilitat dels complexos d'or(III) en condicions catalítiques, també es va trobar que AuCl₃ simple era un catalitzador eficient en alguns casos. Per exemple, Hashmi va informar d'una reacció de Diels-Alder catalitzada per AuCl₃ alquí/furan, un tipus de cicloaddició que no es produeix habitualment, per a la síntesi de fenols 2,3-dissubstituïts:[22]

Estudis mecànics posteriors conclouen que no es tracta d'una transformació concertada, sinó d'una hidroarilació inicial d'alquins seguida d'una sèrie de reordenaments intramoleculars no evidents, que conclouen amb una electrociclització i rearomatització de 6π.
Els efectes relativistes són significatius en la química d'organoor a causa de la gran càrrega nuclear del metall (Z = 79). Com a conseqüència dels orbitals 5d expandits relativistament, el fragment LAu pot estabilitzar un carbocatió veí mitjançant la donació d'electrons a l'orbital buit de tipus p. Així, a més de la seva esperada reactivitat semblant a un carbocatió, aquests cations també presenten un caràcter carbè important, una propietat que s'ha aprofitat en transformacions catalítiques com la ciclopropanació i la inserció de C-H.[23] Els èsters de propargil poden servir com a precursors d'intermedis catiònics d'or-vinilcarbè, que poden reaccionar amb alquens de manera concertada per oferir el producte de ciclopropanació. L'ús d'un lligand quiral ((R)-DTBM-SEGPHOS) va donar lloc a excel·lents nivells d'enantioselectivitat.[24]


Tot i que Echavarren va informar per primera vegada de la preparació de complexos quirals de bisfosfinadior(I) per a la catàlisi enantioselectiva d'or mitjançant el típic mecanisme de pi-activació,[25] Hayashi i Ito van descriure un exemple primerenc i atípic de catàlisi enantioselectiva per or el 1986.[26] En aquest procés, el benzaldehid i l'isocianoacetat de metil pateixen ciclització en presència d'un lligand quiral de ferrocenilfosfina i d'un complex bis(isocianur)or(I) per formar una oxazolina quiral. Com que les oxazolines es poden hidrolitzar per proporcionar un 1,2-aminoalcohol, aquesta reacció constitueix el primer exemple de reacció aldòlica asimètrica i catalítica.


En contrast amb les altres reaccions descrites anteriorment, aquesta reacció no implica l'activació d'un doble o triple enllaç C-C per l'or. En una imatge mecànica simple, l'or(I) es coordina simultàniament amb dos lligands de fosfina i el grup isocianat de carboni que després és atacat pel grup carbonil.[27] Estudis addicionals sobre el mode d'enllaç d'Au(I) indiquen que aquesta senzilla imatge pot haver de ser revisada.
La catàlisi heterogènia d'or és una ciència més antiga. L'or és un metall atractiu per la seva estabilitat contra l'oxidació i la seva varietat en la morfologia, per exemple els materials de clúster d'or. S'ha demostrat que l'or és efectiu en l'oxidació de CO a baixa temperatura i en la hidrocloració de l'acetilè a clorurs de vinil. Es debat la naturalesa exacta del lloc catalític en aquest tipus de procés.[28] La idea que l'or pugui catalitzar una reacció no implica que sigui l'única manera. No obstant això, altres metalls poden fer la mateixa feina de manera econòmica, sobretot en els darrers anys el ferro (vegeu la química organoferro).
Reaccions catalitzades per or[modifica]
Tot i que no té importància comercial, l'or catalitza moltes transformacions orgàniques, normalment la formació d'enllaços carboni-carboni a partir de Au(I) i la formació d'enllaços C-X (X = O, N) a partir de l'estat Au(III), a causa de l'acidesa de Lewis més dura d'aquest ió. Durant l'última dècada, diversos estudis han demostrat que l'or pot catalitzar de manera eficient les reaccions d'acoblament creuat de C-C i C-heteroàtoms que procedeixen a través d'un cicle Au(I) / Au(III).[29] Hong C. Shen va resumir les reaccions homogènies que formen compostos cíclics en 4 categories principals:[30]
- Addició nucleòfila d'heteroàtoms als enllaços C-C insaturats, especialment per formar petits heterocicles (furans, pirrols, tiofenols)
- Hidroarilació: bàsicament una reacció de Friedel-Crafts utilitzant complexos metall-alquí. Per exemple, la reacció del mesitilè amb el fenilacetilè:[31]

- Ciclització enina, en particular cicloisomerització, un dels primers exemples és una cicloisomerització enina 5-exo-dig 1,6:[32]

- reaccions de cicloaddició amb els primers exemples de la cicloaddició d'un òxid de nitril amb un alquí.[33]
Altres reaccions són l'ús d'or en l'activació de l'enllaç C-H[34] i les reaccions aldòliques. L'or també catalitza les reaccions d'acoblament.[35]
Limitacions[modifica]
Tot i que la hidrofuncionalització catalitzada per or d'alquins, al·lens i alcohols al·lílics[36] es produeix fàcilment en condicions relativament suaus, els alquens no activats segueixen sent substrats pobres en la majoria dels casos,[37] en gran part a causa de la resistència dels complexos intermedis alquilor(I) a la protodesauració.[38] El desenvolupament de transformacions intermoleculars catalitzades per or també s'ha quedat endarrerit amb el desenvolupament de les intramoleculars.[39]
Referències[modifica]
- ↑ 1,0 1,1 Elschenbroich i Salzer, 1992.
- ↑ 2,0 2,1 Parish, 1997, p. 55-62.
- ↑ 3,0 3,1 Parish, 1998, p. 14-21.
- ↑ Mathews i Watters, 2002, p. 108-111.
- ↑ Schmidbaur i Schier, 2011, p. 370-412.
- ↑ Wang et al., Li, p. 16368-16370.
- ↑ Kharasch i Isabell, 1931, p. 3053-3059.
- ↑ Rice i Tobias, 1975, p. 2402-2407.
- ↑ Toste, F. Dean «Gold catalysis for organic synthesis» (en anglès). Beilstein Journal of Organic Chemistry.
- ↑ Raubenheimer i Schmidbaur, 2014, p. 2024-2036.
- ↑ Ranieri, Escofet i Echavarren, 2015, p. 7103-7118.
- ↑ Wang, Lackner i Toste, 2013, p. 889-901.
- ↑ Zhdanko i Maier, 2015, p. 5994-6004.
- ↑ Wang et al., Thummanapelli, p. 9012-9019.
- ↑ Homs, Escofet i Echevarren, 2013, p. 5782-5785.
- ↑ Fürstner i Dvies, 2007, p. 3410-3449.
- ↑ Shen, 2008, p. 3885-3903.
- ↑ Norman, Parr i Thomas, 1976, p. 1983.
- ↑ Fukuda i Utimoto, 1991, p. 3729-3731.
- ↑ Teles, Brode i Chabanas, 1998, p. 1415-1418.
- ↑ Nugent, 2012, p. 8936-8949.
- ↑ Hashmi, Frost i Bats, 2000, p. 11553-11554.
- ↑ Gorin i Toste, 2007, p. 395-403.
- ↑ Johansson et al., 2005, p. 18002-18003.
- ↑ Muñoz et al., 2005, p. 1293-1300.
- ↑ Ito, Sawamura i Hayashi, 1986, p. 6405-6406.
- ↑ Togni i Pastor, 1990, p. 1649-1664.
- ↑ Hutchings, Bruts i Schmidbaur, 2008, p. 1759-1765.
- ↑ Nijamudheen i Datta, 2020, p. 1442-1487.
- ↑ Shen, 2008, p. 7847-7870.
- ↑ Reetz i Sommer, 2003, p. 3485-3496.
- ↑ Nieto-Oberhuber et al., Cárdenas, p. 2402-2406.
- ↑ Gasparrini et al., Palmieri, p. 4401-4402.
- ↑ Hoffmann-Röder i Krause, 2005, p. 387-391.
- ↑ Wegner i Auzias, 2011, p. 8236-8247.
- ↑ Bandini, 2011, p. 994-995.
- ↑ Zhang, Lee i Widenhoefer, 2009, p. 5372-5373.
- ↑ LaLonde et al., Kelley, p. 226.
- ↑ Muratore et al., 2004, p. 3066-3082.
Bibliografia[modifica]
- Bandini, Marco «Allylic Alcohols: Sustainable Sources for Catalytic Enantioselective Alkylation Reactions» (en anglès). Angewandte Chemie International Edition, 50(5), febrer 2011. DOI: 10.1002/anie.201006522. ISSN: 1521-3773. PMID: 21268189.
- Elschenbroich, C.; Salzer, A. Organometallics : A Concise Introduction (en anglès). Weinheim: Wiley-VCH, 1992. ISBN 3-527-28165-7.
- Fürstner, A.; Davies, P. W. «Catalytic Carbophilic Activation: Catalysis by Platinum and Gold π Acids» (en anglès). Angewandte Chemie International Edition, 46(19), 2007. DOI: 10.1002/anie.200604335. PMID: 17427893.
- Fukuda, Y.; Utimoto, K. «Effective transformation of unactivated alkynes into ketones or acetals with a gold(III) catalyst» (en anglès). The Journal of Organic Chemistry, 56(11), 1991. DOI: 10.1021/jo00011a058.
- Gasparrini, F.; Giovannoli, M.; Misiti, D.; Natile, G.; Palmieri, G.; Maresca, L. «Gold(III)-catalyzed one-pot synthesis of isoxazoles from terminal alkynes and nitric acid» (en anglès). Journal of the American Chemical Society, 115(10), 1993. DOI: 10.1021/ja00063a084.
- Gorin, David J.; Toste, F. Dean «Relativistic effects in homogeneous gold catalysis» (en anglès). Nature, 446(7134), 2007. Bibcode: 2007Natur.446..395G. DOI: 10.1038/nature05592. PMID: 17377576.
- Hashmi, A. S. K.; Frost, T. M.; Bats, J. W. «Highly Selective Gold-Catalyzed Arene Synthesis» (en anglès). Journal of the American Chemical Society, 122(46), 2000. DOI: 10.1021/ja005570d.
- Hoffmann-Röder, A.; Krause, N. «The golden gate to catalysis» (en anglès). Organic & Biomolecular Chemistry, 3(3), 2005. DOI: 10.1039/b416516k. PMID: 15678171.
- Homs, Anna; Escofet, Imma; Echavarren, Antonio M. «On the Silver Effect and the Formation of Chloride-Bridged Digold Complexes» (en anglès). Organic Letters, 15(22), 2013. DOI: 10.1021/ol402825v. PMC: 3833279. PMID: 24195441.
- Hutchings, G. J.; Brust, M.; Schmidbaur, H. «Gold - an introductory perspective» (en anglès). Chemical Society Reviews, 37(9), 2008. DOI: 10.1039/b810747p. PMID: 18762825.
- Ito, Y.; Sawamura, M.; Hayashi, T. «Catalytic asymmetric aldol reaction: Reaction of aldehydes with isocyanoacetate catalyzed by a chiral ferrocenylphosphine-gold(I) complex» (en anglès). Journal of the American Chemical Society, 108(20), 1986. DOI: 10.1021/ja00280a056.
- Johansson, Magnus J.; Gorin, David J.; Staben, Steven T.; Toste, F. Dean «Gold(I)-Catalyzed Stereoselective Olefin Cyclopropanation» (en anglès). Journal of the American Chemical Society, 127(51), novembre 2005. DOI: 10.1021/ja0552500. PMID: 16366541.
- Kharasch, M. S.; Isbell, Horace S. «The Chemistry of Organic Gold Compounds. III. Direct Introduction of Gold into the Aromatic Nucleus (Preliminary Communication)» (en anglès). Journal of the American Chemical Society, 53(8), agost 1931. DOI: 10.1021/ja01359a030. ISSN: 0002-7863.
- LaLonde, Rebecca L.; Brenzovich, William E. Jr.; Benítez, Diego; Tkatchouk, Ekaterina; Kelley, Kotaro; Goddard, William A. III; Toste, F. Dean «Alkylgold complexes by the intramolecular aminoauration of unactivated alkenes» (en anglès). Chemical Science, 1(2), 2010. DOI: 10.1039/C0SC00255K. PMC: 3866133. PMID: 24358445.
- Mathews, J. A.; Watters, L. L. «The Carbide of Gold» (en anglès). Journal of the American Chemical Society, 22(2), maig 2002. DOI: 10.1021/ja02040a010.
- Muñoz, M. Paz; Adrio, Javier; Carretero, Juan Carlos; Echavarren, Antonio M. «Ligand Effects in Gold- and Platinum-Catalyzed Cyclization of Enynes: Chiral Gold Complexes for Enantioselective Alkoxycyclization» (en anglès). Organometallics, 24(6), febrer 2005. DOI: 10.1021/om0491645.
- Muratore, Michael E.; Homs, Anna; Obradors, Carla; Echavarren, Antonio M. «Meeting the Challenge of Intermolecular Gold(I)-Catalyzed Cycloadditions of Alkynes and Allenes» (en anglès). Chemistry: An Asian Journal, 9(11), novembre 2014. DOI: 10.1002/asia.201402395. ISSN: 1861-471X. PMC: 4676923. PMID: 25048645.
- Nieto-Oberhuber, C.; Muñoz, M. P.; Buñuel, E.; Nevado, C.; Cárdenas, D. J.; Echavarren, A. M. «Cationic Gold(I) Complexes: Highly Alkynophilic Catalysts for theexo- andendo-Cyclization of Enynes» (en anglès). Angewandte Chemie International Edition, 43(18), 2004. DOI: 10.1002/anie.200353207. PMID: 15114573.
- Nijamudheen, A.; Datta, Ayan «Gold‐Catalyzed Cross‐Coupling Reactions: An Overview of Design Strategies, Mechanistic Studies, and Applications» (en anglès). Chemistry: A European Journal, 26(7), 2020. DOI: 10.1002/chem.201903377. PMID: 31657487.
- Norman, R. O. C.; Parr, W. J. E.; Thomas, C. B. «The reactions of alkynes, cyclopropanes, and benzene derivatives with gold(III)» (en anglès). Journal of the Chemical Society, Perkin Transactions 1, 18, 1976. DOI: 10.1039/P19760001983.
- Nugent, W. A. «"Black Swan Events" in Organic Synthesis» (en anglès). Angewandte Chemie International Edition, 51(36), 2012. DOI: 10.1002/anie.201202348. PMID: 22893229.
- Parish, R. V. «Organogold chemistry: II reactions» (en anglès). Gold Bulletin, 30(2), 1997. DOI: 10.1007/BF03214757.
- Parish, R. V. «Organogold chemistry: III applications» (en anglès). Gold Bulletin, 31, 1998. DOI: 10.1007/BF03215470.
- Ranieri, Beatrice; Escofet, Imma; Echavarren, Antonio M. «Anatomy of gold catalysts: facts and myths» (en anglès). Org. Biomol. Chem., 13(26), juny 2015. DOI: 10.1039/c5ob00736d. ISSN: 1477-0539. PMC: 4479959. PMID: 26055272.
- Raubenheimer, H. G.; Schmidbaur, H. «The Late Start and Amazing Upswing in Gold Chemistry» (en anglès). Journal of Chemical Education, 91(12), 2014. Bibcode: 2014JChEd..91.2024R. DOI: 10.1021/ed400782p.
- Reetz, M. T.; Sommer, K. «Gold-Catalyzed Hydroarylation of Alkynes» (en anglès). European Journal of Organic Chemistry, 2003(18), 2003. DOI: 10.1002/ejoc.200300260.
- Rice, Gary W.; Tobias, R. Stuart «Synthesis of tetramethylaurate(III). Structures of lithium dimethylaurate and lithium tetramethylaurate in solution» (en anglès). Inorganic Chemistry, 14(10), octubre 1975. DOI: 10.1021/ic50152a020. ISSN: 0020-1669.
- Schmidbaur, Hubert; Schier, Annette «Aurophilic interactions as a subject of current research: an up-date» (en anglès). Chemical Society Reviews, 41(1), desembre 2011. DOI: 10.1039/C1CS15182G. ISSN: 1460-4744. PMID: 21863191.
- Shen, H. C. «Recent advances in syntheses of heterocycles and carbocycles via homogeneous gold catalysis. Part 1: Heteroatom addition and hydroarylation reactions of alkynes, allenes, and alkenes» (en anglès). Tetrahedron, 64(18), 2008. DOI: 10.1016/j.tet.2008.01.081.
- Shen, H. C. «Recent advances in syntheses of carbocycles and heterocycles via homogeneous gold catalysis. Part 2: Cyclizations and cycloadditions» (en anglès). Tetrahedron, 64(34), 2008. DOI: 10.1016/j.tet.2008.05.082.
- Teles, J. H.; Brode, S.; Chabanas, M. «Cationic Gold(I) Complexes: Highly Efficient Catalysts for the Addition of Alcohols to Alkynes» (en anglès). Angewandte Chemie International Edition, 37(10), 1998. DOI: 10.1002/(SICI)1521-3773(19980605)37:10<1415::AID-ANIE1415>3.0.CO;2-N. PMID: 29710887.
- Togni, A.; Pastor, S. D. «Chiral cooperativity: The nature of the diastereoselective and enantioselective step in the gold(I)-catalyzed aldol reaction utilizing chiral ferrocenylamine ligands» (en anglès). The Journal of Organic Chemistry, 55(5), 1990. DOI: 10.1021/jo00292a046.
- Wang, X. B.; Wang, Y. L.; Yang, J.; Xing, X. P.; Li, J.; Wang, L. S. «Evidence of Significant Covalent Bonding in Au(CN)₂−» (en anglès). Journal of the American Chemical Society, 131(45), 2009. DOI: 10.1021/ja908106e. PMID: 19860420.
- Wang, Dawei; Cai, Rong; Sharma, Sripadh; Jirak, James; Thummanapelli, Sravan K. «"Silver Effect" in Gold(I) Catalysis: An Overlooked Important Factor» (en anglès). Journal of the American Chemical Society, 134(21), maig 2012. DOI: 10.1021/ja303862z. PMID: 22563621.
- Wang, Yi-Ming; Lackner, Aaron D.; Toste, F. Dean «Development of Catalysts and Ligands for Enantioselective Gold Catalysis» (en anglès). Accounts of Chemical Research, 47(2), novembre 2013. DOI: 10.1021/ar400188g. PMC: 3960333. PMID: 24228794.
- Wegner, H. A.; Auzias, M. «Gold for C-C coupling reactions: a Swiss-Army-knife catalyst?» (en anglès). Angewandte Chemie International Edition, 50(36), 2011. DOI: 10.1002/anie.201101603. PMID: 21818831.
- Zhang, Zhibin; Lee, Seong Du; Widenhoefer, Ross A. «Intermolecular Hydroamination of Ethylene and 1-Alkenes with Cyclic Ureas Catalyzed by Achiral and Chiral Gold(I) Complexes» (en anglès). Journal of the American Chemical Society, 131(15), abril 2009. DOI: 10.1021/ja9001162. ISSN: 0002-7863. PMC: 2891684. PMID: 19326908.
- Zhdanko, Alexander; Maier, Martin E. «Explanation of "Silver Effects" in Gold(I)-Catalyzed Hydroalkoxylation of Alkynes» (en anglès). ACS Catalysis, 5(10), setembre 2015. DOI: 10.1021/acscatal.5b01493.
Compostos de carboni amb altres elements de la taula periòdica | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||